SATqPCR FAQ
Frequently asked questions
- How to design an experiment
- How to enter your data
- What are REF genes
- Statistical notation
- Computation of normalization factor
- Normalization of relative quantities
- Computation of relative quantities
- How do we compare samples
- How to rescale the normalized expression for each GOI
- Graphical representation of your analysis
- Result description
How to design an experiment ?
You should at least have 3 biological replicates and at least 2 technical replicates for each sample of your experiments. Each gene you want to quantify should have a validated essay according to the MIQE standard. To follow the MIQE you should have calculated the efficiency of the primers you used to amplify a given sequence. This validation is done using calibration curves with at least 5 dilution points. The efficiency of the PCR reaction should be between 90-100% with an R2 between 0.99-1.Amplicons of different genes can have different efficiency values but their all should be in the range of 90-100%. Once you have validated your amplicon for each gene of your assay you can use them to estimate the expression value of these genes in our experiments. SATqPCR is taking into account the efficiency difference between your GOI. In the text file you provide for the analysis (example.txt) you have to give the efficiency for each gene amplification in the first row.
The design of your experiment is describing the number of biological samples including your biological control, the number of technical replicates, and the number of genes tested (GOI and putative reference genes).
How to enter your data
Input data must have the structure of our example file "example.txt".Please construct your input data according to this data structure:
- samplename_X where X is the number identifying the biological replicate
- all technical replicates of the same biological sample have exactly the same name
- All samples from the same biological replicate have to be in a same block,
each block corresponds to a biological replicate i (i=1,... m, "sample1_i...samplei_i"
This is represented in the table below :
Sample GENE1 GENE2 GENE3 GENE1 GENE2 GENE3 <= one column by gene
efficiency 1.96 1.77 2.2 1.93 1.83 1.87
sample1_1 30.5 30.83 32.77 18.92 32.48 18.37 |
sample1_1 31.08 30.83 33.03 19.46 33.67 19.72 |
sample2_1 30.12 29.76 32.95 15.73 32.9 18.91 | Biological replicate 1
sample2_1 30.44 30.08 32.45 15.82 32.27 18.91 |
sampleN_1 30.68 30.37 33.24 17.56 31.87 20.16 |
sampleN_1 31.63 30.73 33.64 17.21 32.46 20.01 |
sample1_2 30.5 29.11 31.62 19.84 34.49 27.22 |
sample1_2 30 29.19 32.08 19.74 32.56 27.11 |
sample2_2 30.97 30.15 33.4 18.09 35.13 28.28 | Biological replicate 2
sample2_2 30.89 30.04 33.73 17.4 34.86 28.25 |
sampleN_2 31.56 30.5 34.2 18.95 32.53 23.53 |
sampleN_2 31.48 30.88 33.9 18.16 32.11 23.32 |
sample1_m 31.07 30.41 32.56 22.11 34.66 30.02 |
sample1_m 31.84 30.84 33.98 21.91 34.67 29.94 |
sample2_m 31.27 30.5 33.16 17.91 32.98 28.44 | Biological replicate m
sample2_m 31.77 30.73 33.72 17.63 33.92 28.17 |
sampleN_m 31.5 31.77 34.51 19.61 33.82 29.42 |
sampleN_m 32.43 32 34.33 19.01 32.68 29.3 |The missing values must be replaced by 'NA' value. The number of technical replications must be identical across all samples. The missing technical replication of a given sample should be added by a row where all values are 'NA' value. Your sample names and your gene names should not have a “space” or “.” or “,” or “;” or “$” or “\” or “/”. Your sample names and your gene names should not start with a number. Your data should be in a text file with a tabular separator.
What are REF genes ?
REFs are the most stably expressed genes across all tested samples and all biological replicates of one data set. Their identification is based on the stability parameter and coefficient of variation. SATqPCR uses the algorithm described in (Vandesompele et al. Genome Biology (2002) 3 (7):0034 1-11 ) to identify the reference genes. The REFs are specific to your studies and should be validated for each data set you want to analyze.You can provide a list of reference genes by yourself but we DO NOT RECOMMEND it. Otherwise, SATqPCR will provide a list of reference genes for you by finding the most stables genes (at least 2).
Statistical notation
m number of biological replicates (index i)
n number of technical replicates (index j)
r number of reference genes (index ??)
Ej PCR amplification efficiency coefficient of gene j
k number of samples
Cq quantitative cycle
QCq relative quantity
SD standard deviation
SE standard error
NF normalization factor
M degree of freedomComputation of normalization factor ?
The normalization factor of sample k in biological replicate i across r reference genes, noted NFik, is given by the geometric mean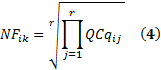
And the standard deviation of this normalization factor is
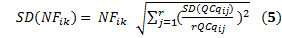 with r corresponding to the number of reference genes.
with r corresponding to the number of reference genes.
Normalization of relative quantities
For each biological replicate i,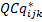 is the normalization of relative quantity of GOI j for each sample k and is calculated by dividing the raw GOI quantity (2) by the normalization factor (4) as follows
is the normalization of relative quantity of GOI j for each sample k and is calculated by dividing the raw GOI quantity (2) by the normalization factor (4) as follows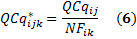
and the standard deviation of this normalized GOI expression is given by
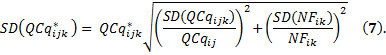
The standard error quantifies scatter. To assess the precision of the mean calculation, we also provide the standard error (SE) value. The confidence interval defined by the measured mean ±1.96 times the SE has a 95% chance of containing the true mean
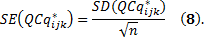
Now, the average of expression level of GOI j for each sample k across m biological replicates is calculated by
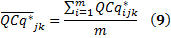
and the SD and SE are given by
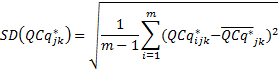
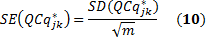
Finally, the user can use the reference sample k,
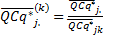
or the smallest sample for rescaling the expression level
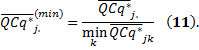
The SD and SE corresponding to the transformation in (11) are
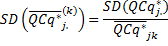
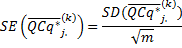
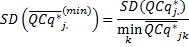
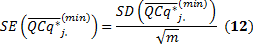
Computation of relative quantities
For each biological replicate i, the average of Cq is computed for n technical replicates of the same gene j: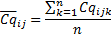
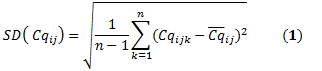
The relative quantity associated with
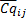 is expressed as follows where the highest expression level set to one,
is expressed as follows where the highest expression level set to one,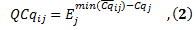
and the standard deviation of QCqij is obtained from (2):
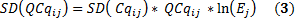
How do we compare samples ?
Comparison between samples is performed using Statistical Significant Test. The user can performed either a t-test or an ANOVA.
For each GOI, all pairs of samples across all biological replicates are compared. Here we used the classical method of pairwise t-tests with a pooled standard deviation. We assumed that the two sample sizes are equal and the two distributions have the same variance. In order to apply the t-test, we have to ensure that the test samples follow Gaussian distributions. Here we use a simple logarithm transformation to transform the normalized relative expression,
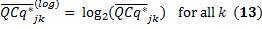
Now, the t statistic to test whether the means are different between the samples S_i and S_j follows a Student's t-distribution with M-1 degrees of freedom and can be calculated as follows:
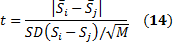
where SD(Si-Sj) is the standard deviation of the differences between the Si and the Sj:
Once a t value is determined, the p-value of the test can be calculated from Student's t-distribution with M-1 degrees of freedom. The user can choose a threshold for the statistical significance to reject or accept the null hypothesis (H0: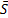 i=j) in favor of the alternative hypothesis (Ha: i≠j).
i=j) in favor of the alternative hypothesis (Ha: i≠j).
Now, the t statistic to test whether the means are different between the samples S_i and S_j follows a Student's t-distribution with M-1 degrees of freedom and can be calculated as follows:
where SD(Si-Sj) is the standard deviation of the differences between the Si and the Sj:
Once a t value is determined, the p-value of the test can be calculated from Student's t-distribution with M-1 degrees of freedom. The user can choose a threshold for the statistical significance to reject or accept the null hypothesis (H0:
i=j) in favor of the alternative hypothesis (Ha: i≠j).
You should give the ANOVA factors associated to your experimental design in a text file. See factor.txt example.
Assumption: normal distributions and independence of the observations
Total Variability= variability explained by the factors + residual variability
The factor.txt file should contain the different factors associated for each sample :
Assumption: normal distributions and independence of the observations
Total Variability= variability explained by the factors + residual variability
The factor.txt file should contain the different factors associated for each sample :
Sample Factor1 Factor2 Factor3 FactorN <= one column by factor
sample1 info1 info2 info3 infoN |
sample2 info1 info2 info3 infoN | <= one row by sample
sampleN info1 info2 info3 infoN |Sample Genotypes Time Tissues
col0h1 Columbia0 1h leaf
col2h1 Columbia0 2h leaf
col4h1 Columbia0 3h leaf
M0h1 Mutant1 1h leaf
M2h1 Mutant1 2h leaf
M4h1 Mutant1 3h leafFactor1, Genotypes, has two modalities: Columbia0 and Mutant1.
Factor2, Time Course, has 3 modalities 1h, 2h and 4h.
Factor3, Tissues, has 2 modalities: tissues and leaf. Etc.
How to rescale the normalized expression for each GOI
Rescaling by minimal sample value
By default, for each gene, your data are rescaled using the sample with the lowest expression.Rescaling by a specific sample
You can rescale your expression data to a chosen sample. You will have to choose the sample name in the box "Sample name used to rescal"Graphical representation of your analysis
SATqPCR will plot rescaled expression for each GOI:- By default, SATqPCR plots all genes and all samples. We will have a figure per gene (all the samples expression values for one gene) and a figure per sample (all the gene expression values for one sample).
- You can select the genes you want to plot across all samples
- You can select the samples you want to plot across all genes
- You can plot the gene expressions with either their standard deviations or with their standard errors (SE). Remember: There is a 95% chance that the confidence interval defined by the measured mean ±1.96 times the SE contains the true mean.
Result description
- If you chose a t-test
SATqPCR returns the following TABLES
'rescaled.expression.txt': all GOI rescaled expression values
'sd.rescaled.expression.txt': standard deviations for all GOI rescaled expression
'se.rescaled.expression.txt': standard errors for all GOI rescaled expression.
'sign.test.gene_GOI': result of the t-test between samples for a GOI. SATqPCR returns for each GOI a sign.test.gene_GOI table.
'sd.rescaled.expression.txt': standard deviations for all GOI rescaled expression
'se.rescaled.expression.txt': standard errors for all GOI rescaled expression.
'sign.test.gene_GOI': result of the t-test between samples for a GOI. SATqPCR returns for each GOI a sign.test.gene_GOI table.
SATqPCR returns the following plot for each GOI and each sample
Gene_GOI: normalized expression level with either SD or SE for all samples
Sample_samplei: normalized expression level with either SD or SE for all GOIs
Sample_samplei: normalized expression level with either SD or SE for all GOIs
- If you chose the ANOVA
SATqPCR returns for one-way ANOVA: aov_GOI_factorx
| Factor1 | residual | ||
| Sum of Squares | 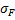 |
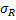 |
|
| Deg. of Freedom | p-1 | n-p | |
| Residual standard error: | # | ||
p : number of modality for factor1
n : number of samplei
SATqPCR returns for two-way ANOVA: aov_GOI_factorx
| Factor1 | Factor2 | residual | ||
| Sum of Squares | 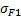 | 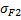 | | |
| Deg. of Freedom | p1-1 | p2-1 | n-p1-p2 | |
| Residual standard error: | # | |||
p1 : number of modalities for factor1
p2 : number of modalities for factor2
n : number of samplei
SATqPCR returns for one-way ANOVA or two-way ANOVA
![]() tukeyTEST_GOI_factorx
tukeyTEST_GOI_factorx
For each factor SATqPCR returns a table as follow
This output indicates that the differences C-A and C-B are significant, while B-A is not significant
| diff | lwr | upr | padj | |
| B-A | -0.06666667 | -1.803101 | 1.669768 | 0.9950379 |
| C-A | -3.80000000 | -5.536435 | -2.063565 | 0.0000260 |
| C-B | -3.73333333 | -5.469768 | -1.996899 | 0.0000337 |
diff : difference between the estimated means of compared modalitiesA, B, C are all the modalities of factorx.
lwr : lower bound of the confidence interval estimated for diff
upr : upper bound of the confidence interval estimated for diff
padj : p value for multiple comparisons (Tukey's Honest Significant Difference method)
This output indicates that the differences C-A and C-B are significant, while B-A is not significant
![]() Normalized_Data_GOI
Normalized_Data_GOI
One for each GOI
| Samples | factorx | Replicat | Value |
| Sample1 | A | 1 | |
| Sample1 | B | 1 | |
| Sample2 | A | 2 | |
| Sample2 | B | 2 | |
| Samplei | # | # |
SATqPCR returns the following plot for each GOI and each sample
- Gene_GOI: normalized expression level with either SD or SE for all samples
- Sample_samplei: normalized expression level with either SD or SE for all GOIs
- Sample_samplei: normalized expression level with either SD or SE for all GOIs